prm-PASEF®
prm-PASEF®
The prm-PASEF® acquisition method has been developed to translate the advantages of the parallel accumulation serial fragmentation (PASEF®) acquisition strategy to the targeted proteomics field.
Abstract
In comparison with standard selected and parallel reaction monitoring (SRM and PRM), the prm-PASEF® increases the number of peptides that can be targeted in a single acquisition method, without compromising the selectivity or the sensitivity. Using a prototype acquisition software, we targeted 201 isotope-labeled synthetic peptides (AQUA peptides) spiked into a 100 ng human HeLa cell line digest. The prm-PASEF® excelled with limits of quantification of 17,2 amol for some peptides utilizing an acquisition method that can monitor 216 precursors over a 30 min LC gradient. The new prm-PASEF® method has a high reproducibility between injections and enables accurate quantification.
Authors
Antoine Lesur1, Pierre-Olivier Schmit2, Christopher Adams3, Gunnar Dittmar1;
1 LIH, Strassen, Luxembourg; 2 Bruker France S.A. Wissembourg, France; 3 Bruker Daltonics Inc, San Jose, USA
Keywords:
4D-Proteomics™, prm-PASEF®, timsTOF Pro, PASEF®, LFQ, absolute quantitation, targeted proteomics
Introduction
Targeted mass spectrometry is a powerful technique to support hypothesis-driven proteomics experiments, for instance, the verification of biomarker candidates in large sample cohorts. It alleviates the problem of missing values between samples but also increases the overall sensitivity compared to data-dependent (DDA) and data-independent (DIA) acquisition methods. Targeted proteomics uses synthetic peptides as an internal standard to normalize the MS signal and confirms the detection of the endogenous peptides with maximum confidence.
A major limitation of targeted proteomics is the compromise that must be found between the number of targets measured in a single run, the duration of the liquid chromatography separation, and the overall sensitivity. This still holds true with the current generation of targeted acquisition methods including SRM and PRM. Therefore, it is only possible to achieve full data completeness for a large number of targeted peptides by either employing longer chromatographic separation or by compromising on the MS sensitivity and selectivity. Alternative approaches like DIA can partially address this issue and rely on a systematic co-isolation and co-*fragmentation of eluting peptides using broad m/z isolation windows. This approach measures most of the peptides, but with limited specificity and sensitivity (all fragments are mixed together) during the achievable MS cycles (the scanning speed of the analyzer has to be minimized) and is not easily combined with short chromatography gradients.
In this application note, we present the prm-PASEF® method, an innovative implementation of the PASEF® acquisition strategy that overcomes the constraints associated with the current targeted proteomics techniques.
Methods
A Human cell line digest (100 ng/µL) was spiked with 201 heavy aqua peptides and 15 light synthetic peptides as described in Figure 1. A dilution curve of nine concentration points ranging from 5.5 to 50,000 amol/µl was generated using 15 heavy AQUA peptides and their light counterpart. This set of peptides served as the internal standard. The other 186 AQUA peptides were spiked at the constant concentration of 2 fmol/µl in all samples.
Samples and controls were injected in technical triplicate and separated on a nano HPLC (nanoElute®, Bruker Daltonics) using 250 mm pulled emitter columns (IonOpticks, Australia) with a 30 min gradient stepped gradient ranging from 2-30% acetonitrile. Peptides were analyzed on a timsTOF Pro mass spectrometer (Bruker Daltonics) operated in prm-PASEF® mode. A single prm-PASEF® method was used for this study: the tims accumulation time was fixed to 50 ms while the tims separation duration was set to 100 ms. The range of mobility values was 0.65-1.3 1/K0 and the covered m/z range was 100-1700 m/z. Data were processed with Skyline-daily (20.1.1.83).
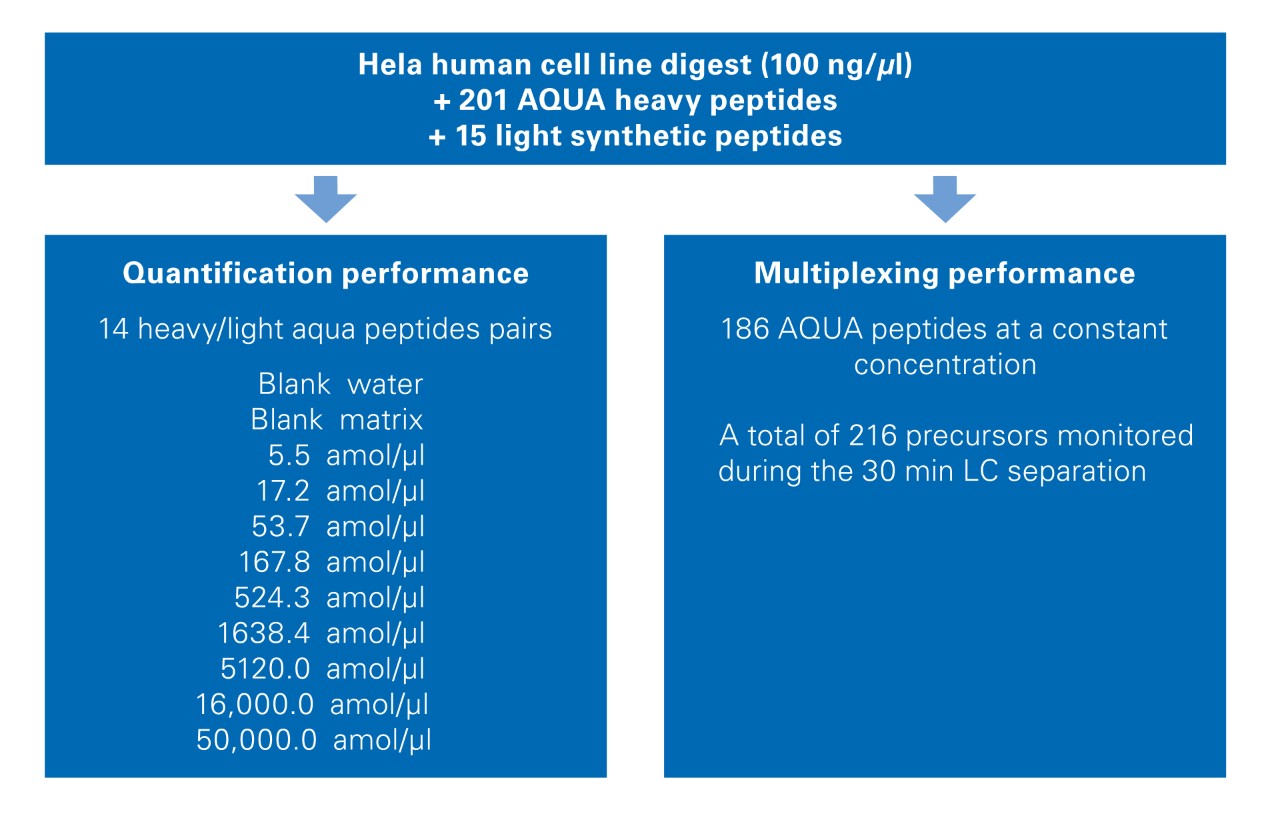
The quantification performance was evaluated measuring the 15 heavy/light ratios at each concentration level and by determining the respective concentration accuracy and relative standard deviation. As a quality control, the concentrations of two curves were back-calculated with the linear regression of the first curve. Limits of quantification (LOQ) were defined as the lowest concentration point within 80% < accuracy < 120% associated with a signal higher than the mean (blanks)+3×SD(blanks).
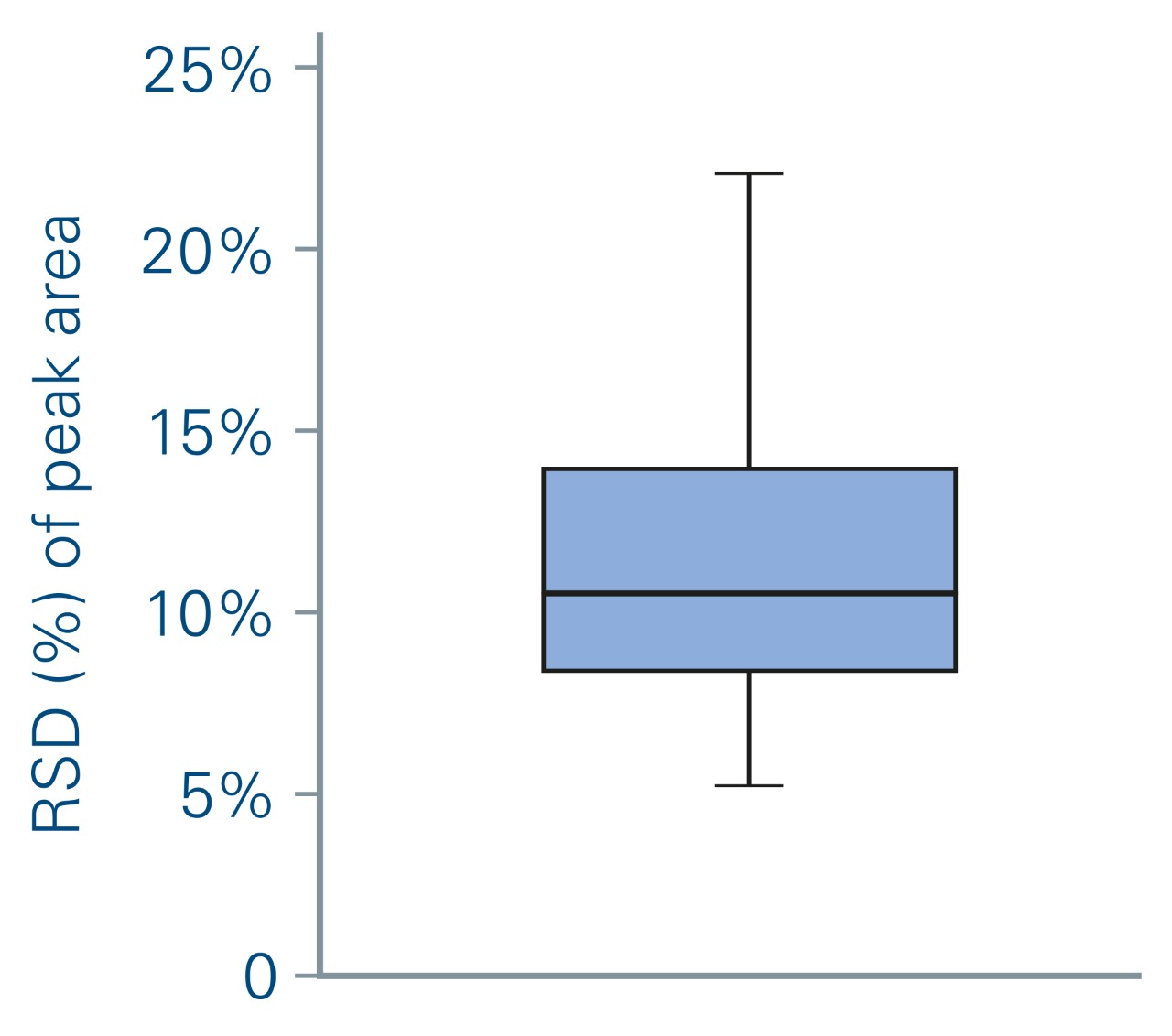
The multiplexing performance was evaluated by measuring the areas of the prm-PASEF® traces for the 186 heavy AQUA peptides spiked at a constant concentration. The relative standard deviation (RSD) of the areas and the number of data points across the chromatographic peak (using the highest PRM transition) was calculated over the 30 prm-PASEF® runs.
Results and Discussion
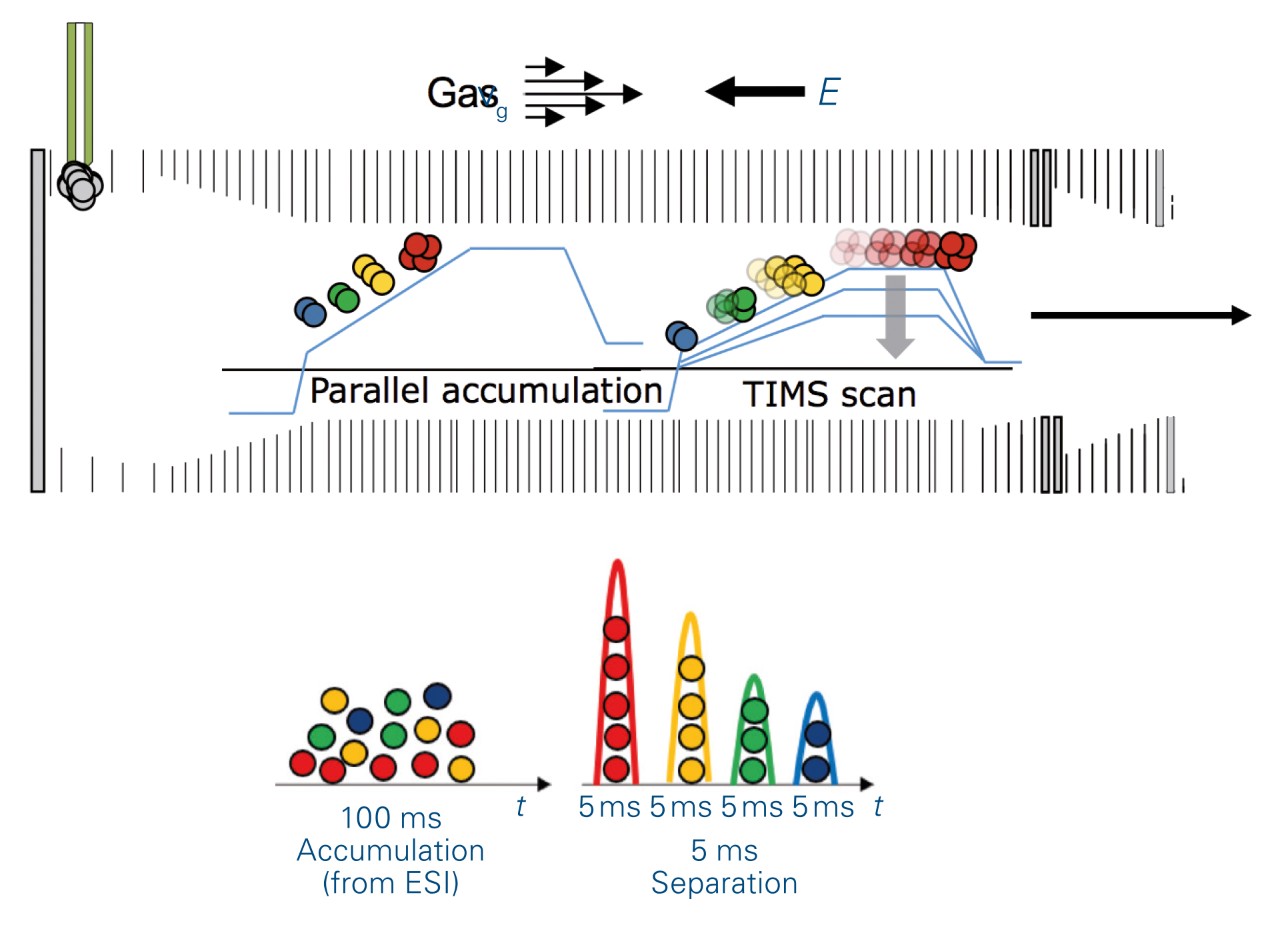
prm-PASEF® takes advantage of the trapped ion mobility spectrometry (tims) device to dramatically increase the multiplexing capability of the method. This allowed for massive parallelization of targeted acquisitions with no detrimental effect on the sensitivity and greater flexibility of implementation with fast LC and UHPLC. The capture and elution principle of the tims cell (Figure 2) allows all the targeted peptides, that sequentially elute in the ion mobility dimension, to be acquired during a 100 ms tims scan event. In addition, the time focusing effect increases the sensitivity because peptides accumulated for 100 ms are then eluted in 5 ms peaks to the MS.
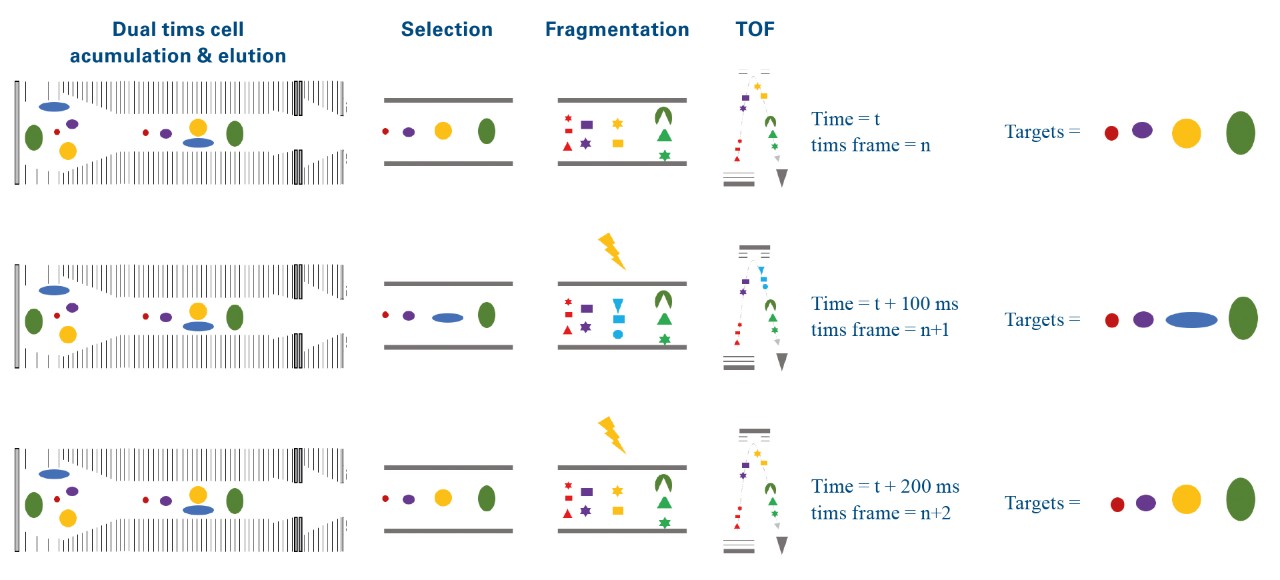
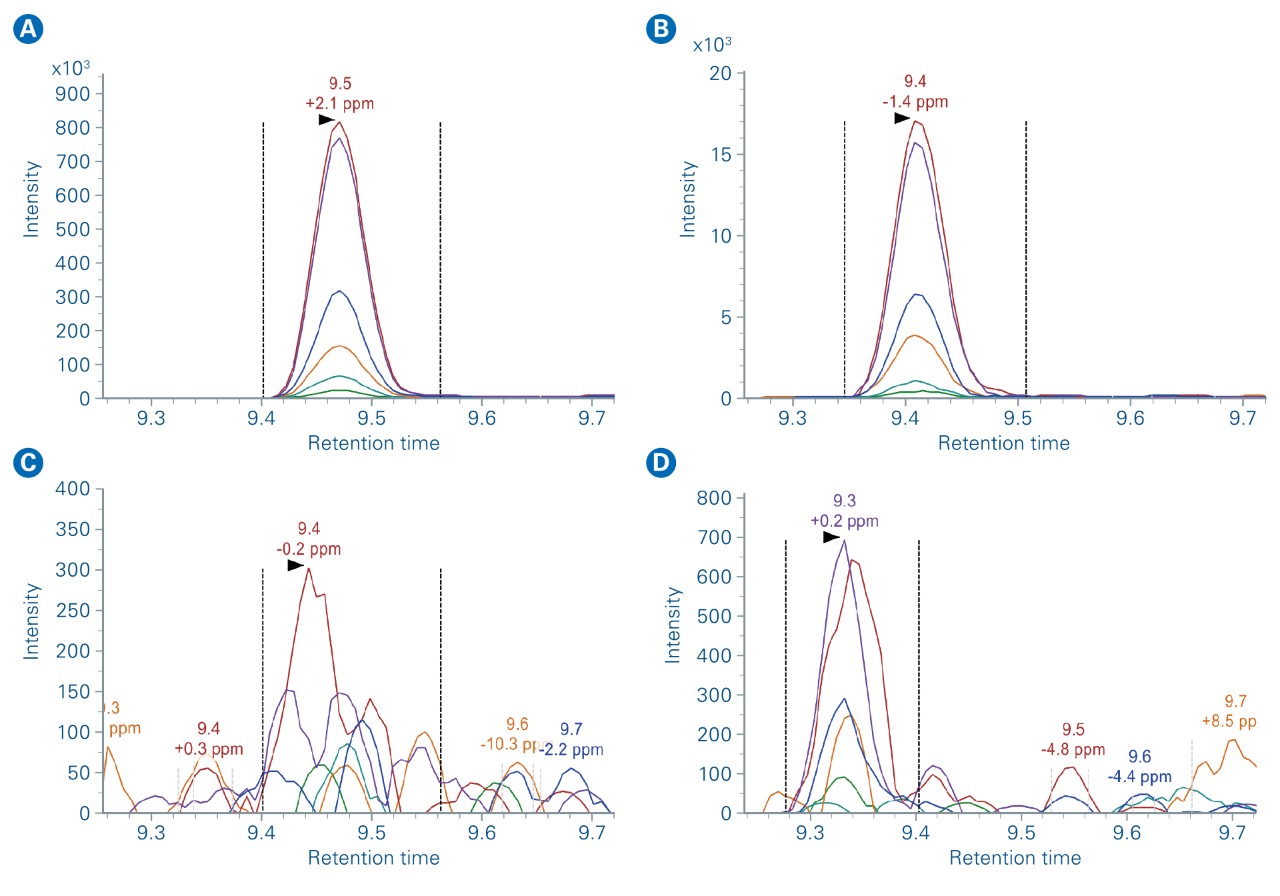
The elution times of the 216 precursors spread over 22 min on a 30 min gradient. In prm-PASEF® the acquisitionwindows are three-dimensional and an isolation window must be defined according to chromatographic elution time, ion mobility, and the quadrupole isolation m/z. As we set the retention windows to 40 s per precursor, it generated substantial overlapping on the LC retention time dimension. However, the IMS dimension reduces the final acquisition window overlap as shown in Figure 5a. Precursor ions with overlapping acquisition windows, which can still be separated in the ion mobility dimension are measured within the same prm-PASEF® frame (Figure 3).
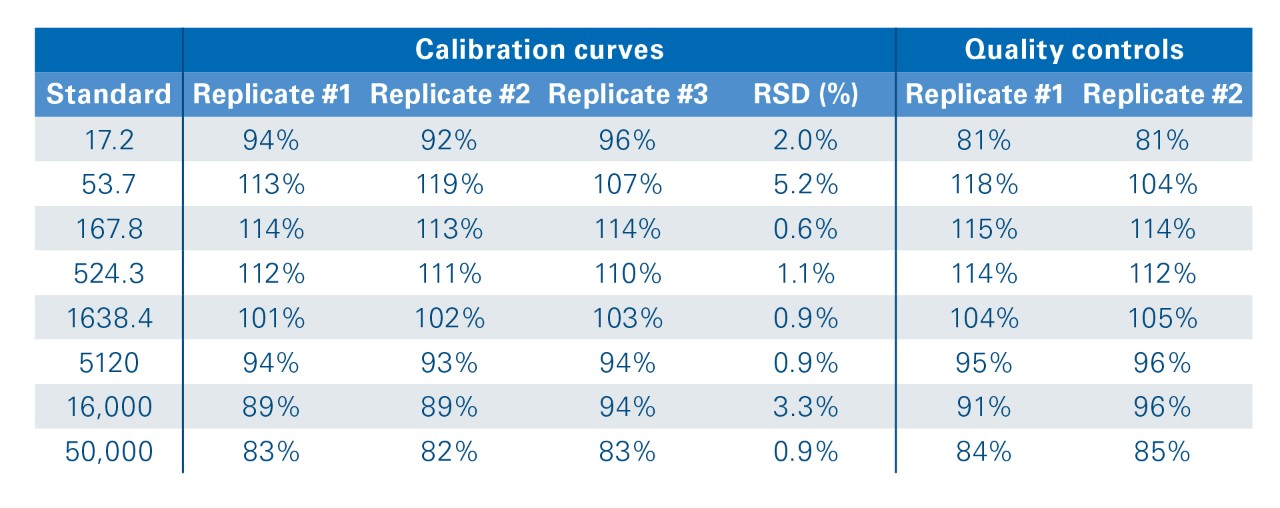
The absolute quantitation potential of prm-PASEF® was evaluated by studying heavy/light ratios for the 15 spiked-in peptides pairs. The combination of selectivity and sensitivity allowed for good signal detection, even at low concentrations (Figure 7). The detailed analysis of the ATVVYQGER heavy/light pair revealed that the signal response can be fitted by linear regression over a concentration factor of 2900 (from 17.2 to 50,000 amol injected column). Moreover, the back-calculation of the concentration for two of the triplicates, based on the calibration curve established with the first one, confirmed that the quantitation can be determined with a ± 20% accuracy at all concentrations (Table 1). The Limit of Quantification (LOQ) was defined as the lowest concentration point within 80% < accuracy < 120% associated with a signal higher than the mean (blanks) + 3X SD(blanks).
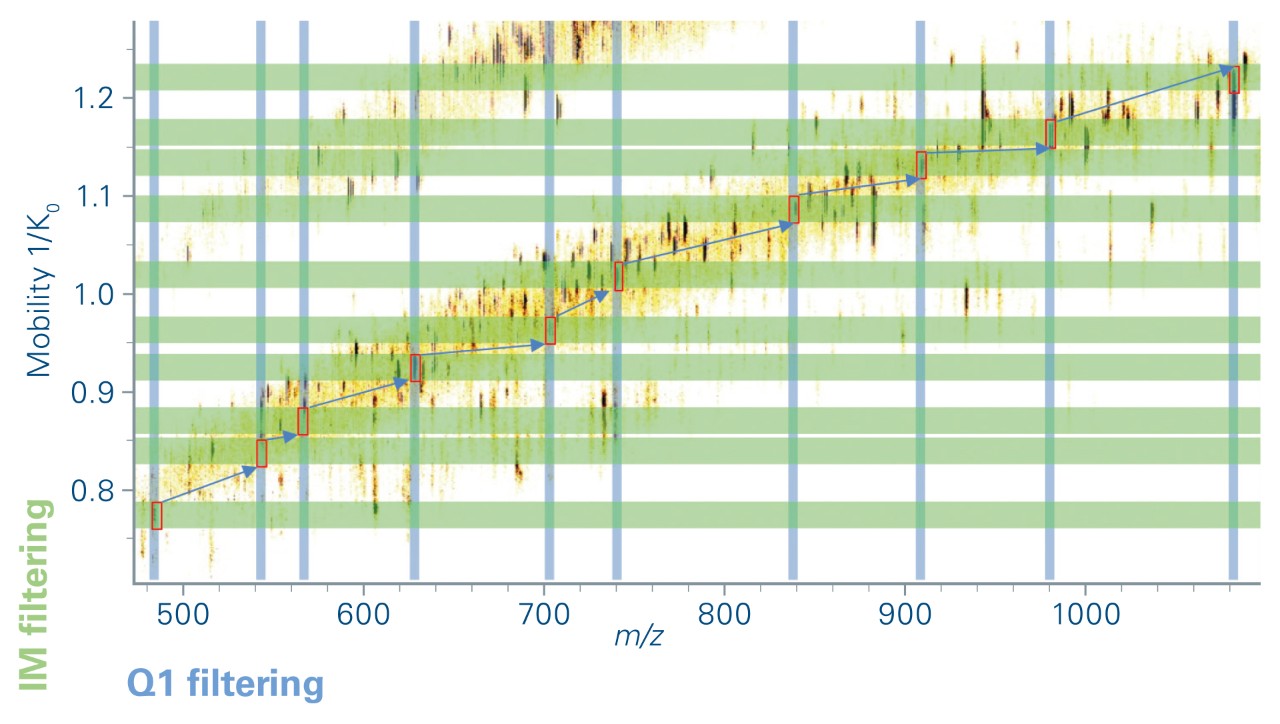
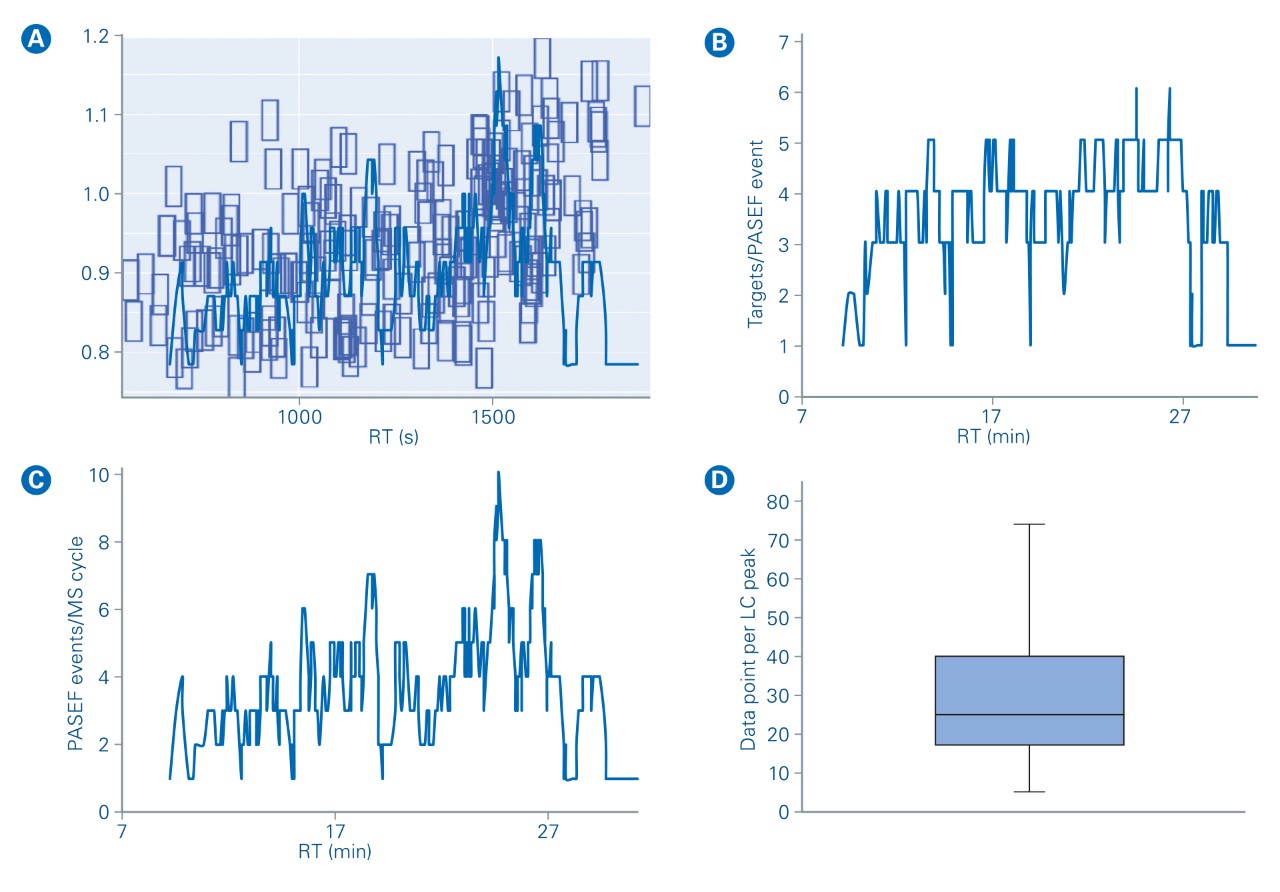
During this experiment, an average of 4 targeted ions was measured within each prm-PASEF® frame (Figure 5b) without affecting the sensitivity nor the cycle time. When several compounds were overlapping in both IMS and LC dimensions, they were measured in successive PASEF® frames (figure 4). On average, there were 4 distinct prm-PASEF® frames in a prm-PASEF® cycle. At the highest precursor density, up to 10 frames were used per MS cycle (Figure 5c). The number of frames per MS cycle does not impact the sensitivity but the number of data points that define the chromatographic peak. Despite this, there was no under-sampling of chromatographic peaks (Figure 5d), the median number of data points per chromatographic peak was 25, suggesting that the experiment could be performed with an even shorter gradient. Good peak sampling and preserved sensitivity allowed measuring all peak areas with a correct RSD even without internal standard normalization (Figure 6).
Conclusion
We established the prm-PASEF® method as the new application for the timsTOF Pro that fully takes advantage of the trapped ion mobility technology. Combining sensitivity, speed, and selectivity, the prm-PASEF® acquisition method delivered high reproducibility and accurate quantitation for either a high number of targets or for application with short chromatography gradients (i.e. 5 min). This makes this methodology particularly promising for clinical targeted proteomics experiments, where a list of peptide markers must be quantified with high accuracy and robustness in large sample cohorts.
For Research Use Only. Not for use in clinical diagnostic procedures.