
Proteoforms
Unlocking another dimension by profiling intact proteoforms
Directly profiling the undigested proteoforms can overcome many of the limitations and constitutes a very powerful tool for biomarker candidate discovery. Bruker’s UHR-QTOFs have the capacity to deal with very complex mixtures of intact proteins with no resolution loss, accuracy or dynamic range making them particularly suited to this task.
Enabling top-down based discovery for clinical research
The complex mixtures of intact proteins in top down proteomics represent a significant challenge for many mass spectrometers. This has been somewhat overcome by the use of extensive pre-fractionation before MS analysis. Despite its effectiveness, pre-fractionation leads to issues with reproducibility when comparing large sample cohorts. Bruker’s UHR-QTOF systems deliver highly reproducible and accurate information from complex mixtures of proteoforms, and require less pre-fractionation of the sample resulting in improved reproducibility and making them compatible with the large sample cohorts that are required for clinical research.
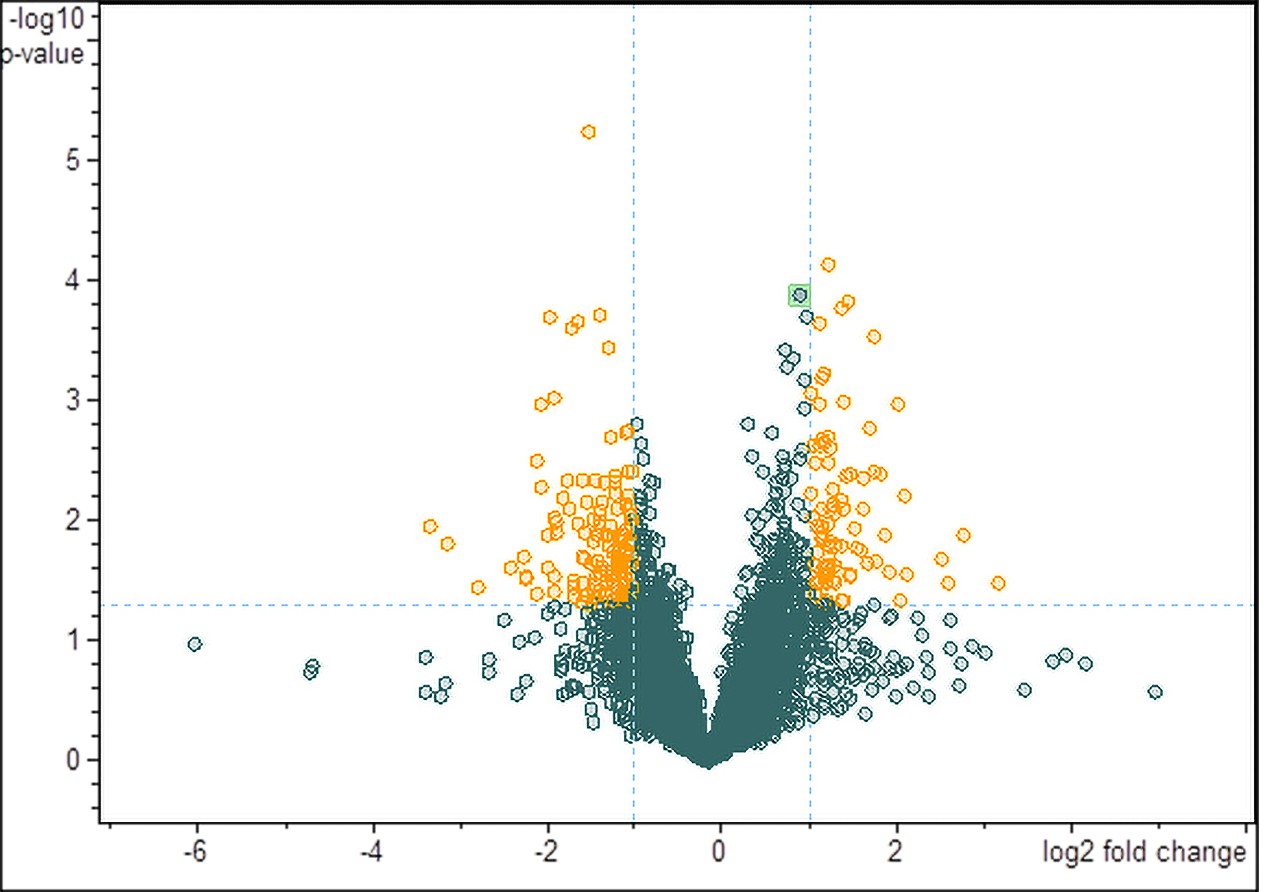
Dealing with intact protein mixtures
Bruker’s UHR-QTOF systems combine Full Sensitivity Resolution with the True Isotopic Pattern (TIP™) for the accurate measurement of intact proteins in complex mixtures. Powerful Dissect™ and SNAP™ algorithms extract the accurate mass and intensity information for a direct label free analysis of intact proteins. All Bruker QTOF instruments deliver quadrupole
CID fragmentation spectra that are well suited to the identification of low to medium MW proteoforms, while the maXis II’s additional ETD fragmentation capabilities generate rich sequence information from even large proteoforms.
The proteoform profiling solution
The proteoform profiling solution from Bruker includes methods that automate the entire process from calibration, extraction and export of the measured intact proteins features to identifying regulated proteofoms through statistical analysis. This information can then be used to create a scheduled precursor fragmentation list to enable efficient fragmentation of the corresponding proteoforms, leading to their identification.






For Research Use Only. Not for use in clinical diagnostic procedures.