Developments and Emerging Trends in Solid-State NMR of Pharmaceuticals
Frederick G. Vogt*
Product Development, GlaxoSmithKline plc.,709 Swedeland Road, King of Prussia, PA 19406, USA
*Present affiliation: Iovance Biotherapeutics, Inc.
The development and application of solid-state NMR spectroscopy (SSNMR) in pharmaceutical analysis continues to progress through research in both academic and industrial settings, particularly as laboratories continue to implement and extend recent accomplishments in the wider field of SSNMR {1, 2}.
In particular, high static field strengths, experiments based on homonuclear dipolar decoupling sequences, and experiments based on dynamic nuclear polarization (DNP) have seen increasing usage in applications to solid pharmaceutical materials and organic materials with similar properties {1-5}.
These methods and other recent developments have enabled access to previously underutilized nuclei, such as 1H, 14N, 17O, and 35Cl, which are of common interest in pharmaceuticals, and has also allowed for improved sensitivity in studies of a variety of crystalline and amorphous phases {6-8}. Both magic-angle spinning (MAS) and static methods continue to be of interest in these applications.
This article highlights two recently-developed approaches with broad potential applicability to pharmaceutical studies involving (1) 1H SSNMR at high fields and with homonuclear dipolar decoupling and (2) DNP enhancement of pharmaceutical SSNMR spectra, for example using amorphous solid dispersions, a common solid form used in drug delivery.
High Field 1H SSNMR and Homonuclear Dipolar Decoupling
The use of high static fields for 1H SSNMR enables detailed
observation of effects such as hydrogen bonding in pharmaceutical
materials of interest. This is particularly relevant in studies of
pharmaceutical cocrystals, which generally involve a molecular complex
of two or more components that when separate are also solid phases.
The great variety of cocrystals that can be formed with a particular drug provides flexibility and in some cases the ability to tailor the physical properties of the resulting cocrystalline phase, and provides new possibilities for drug molecules that are not amenable to the formation of salts.
For example, Fig. 1 illustrates the 1H spectra obtained at a static field of 16.4 T and at a MAS rate of 35 kHz for four cocrystals of the drug tenoxicam {9}. The spectra were obtained using a 2.5 mm double-resonance probe that also enables observation of heteronuclei with a lower gyromagnetic ratio, such as 14N, 17O, and 35Cl,. The spectra offer relatively high resolution and allow for observation of 1H signals for many proton positions of interest {9}.
The spectral region from approximately 9 ppm to 15 ppm is commonly populated by resonances assigned to hydrogen bonding protons, which are usually of significance in cocrystal studies because of the strong influence of hydrogen bonding within the crystal structures of these phases.
The use of high field 1H SSNMR experiments can enable rapid assessment of the success of the formation of a cocrystal along while also providing information on the resulting hydrogen bonding trends, all without the need to obtain a crystal structure.
In addition to higher field strengths, the use of homonuclear dipolar decoupling in the observation of 1H spectra also offers many benefits for studies of pharmaceutical materials.
Many pharmaceutical materials of interest are amorphous and lack long range order, which limits the applicability of studies using diffraction methods. These systems can still be beneficially studied using 1H SSNMR.
In Fig. 2, the 1H SSNMR spectrum of amorphous quinapril hydrochloride obtained using MAS alone at 16.4 T is compared to the 1H spectrum obtained using the windowed eDUMBO-122 pulse sequence for homonuclear 1H decoupling at the same field.
In the case of amorphous quinapril hydrochloride, the use of DUMBO decoupling reveals the presence of at least two proton resonances in the hydrogen bonding region of the spectrum, which are likely related to the carboxylic acid and protonated amine groups in the amorphous solid.
Even without detailed interpretation, these observations highlight the ability of SSNMR to probe structure in amorphous pharmaceutical materials. The superior resolution available from the use of high fields and homonuclear dipolar decoupling enables detailed assignments of 1H spectra obtained directly and through 1H-1H, 1H-13C, and other 2D correlation methods with or without the availability of a crystal structure and an accompanying DFT chemical shielding calculation {1, 2}.
Dynamic Nuclear Polarization
The use of DNP methods has been shown to allow for significant
signal enhancements in studies of microcrystalline solids with longer 1H T1
values {4} and in an amorphous organic molecule {5}. Recent
developments in DNP instrumentation and in stable radical molecules have
further extended the utility of DNP methods {10}.
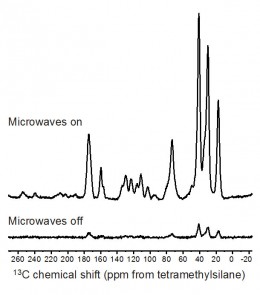
An example of the potential of DNP is shown in Fig. 3 for an amorphous solid dispersion (also known as a glass solution) containing 30% w/w of the drug diflunisal in the polymer polyvinylpyrrolidone (PVP) {7}. A significant enhancement is observed by comparison of the cross-polarization (CP) MAS spectra in Fig. 3, which were obtained at 100 K using microwave irradiation with a 263 GHz continuous-wave gyrotron source, microwave transmission line, 3.2 mm low temperature MAS probe, and 400 MHz Bruker Avance III wide-bore SSNMR spectrometer {10}.
Strong signals from the amorphous drug are seen e.g. in the 140 to 100 ppm region after 16 scans with microwave irradiation, highlighting the potential for both fast 1D analysis and improved sensitivity for 2D experiments. Amorphous solid dispersions represent an area of intense interest in pharmaceutical development, and the ability of DNP to enhance the signal of 13C nuclei and other low sensitivity nuclei should enable greater structural information to be obtained from these systems.
Future Directions
The examples described here represent just a small portion of the recent
developments in SSNMR spectroscopy, which also feature increased access
to quadrupolar nuclei, greater use of paramagnetic probes and isotope
labeling, improved computational methods for spectral prediction and
analysis, and increased application of multidimensional methods.
Translation of these methods for use with pharmaceutical materials has provided more insight into the structure and ultimately the performance of these materials. As advances in SSNMR methods continue, the adaptation of these developments for use with pharmaceutical systems is also expected to continue.
Acknowledgements
Dr. Shane Pawsey and Dr. Jochem Struppe (Bruker Biospin) are thanked for experimental assistance and for providing data.
References
1. Geppi, M.; Mollica, G.; Borsacchi, S.; Veracini, C. A. Appl.
Spectrosc. Rev. 2008, 43, 202-302.
2. Vogt, F. G. Fut. Med. Chem., 2010,
2, 915-921.
3. Madhu, P. K. Solid State Nucl. Magn. Reson. 2009, 35,
2-11.
4. Rossini, A. J.; Zagdoun, A.; Hegner, F.; Schwarzwalder, M.;
Gajan, D.; Coperet, C.; Lesage, A.; Emsley, L. J. Am. Chem. Soc. 2012,
134, 16899-16908.
5. Ong, T. C.; Mak-Jurkauskas, M. L.; Walish, J. J.;
Michaelis, V. K.; Corzilius, B.; Smith, A. A.; Clausen, A. M.; Cheetham,
J. C.; Swager, T. M.; Griffin, R. G. J. Phys. Chem. B, 2013, 117,
3040-3046.
6. Tatton, A. S.; Pham, T. N.; Vogt, F. G.; Iuga, D.;
Edwards, A. J.; Brown, S. P. Mol. Pharm. 2013, 10, 999-1007.
7. Vogt, F.
G.; Yin, H.; Forcino, R. G.; Wu, L. Mol. Pharm. 2013, 10, 3433-3446
8.
Hamaed, H.; Pawlowski, J. M.; Cooper, B. F. T.; Fu, R. Q.; Eichhorn, S.
H.; Schurko, R. W. J. Am. Chem. Soc. 2008, 130, 11056-11065.
9. Patel,
J. R.; Carlton, R. A.; Needham, T. E.; Chichester, C. O.; Vogt, F. G.
Int. J. Pharm. 2012, 436, 685-706.
10. Rosay, M.; Tometich, L.; Pawsey,
S.; Bader, R.; Schauwecker, R.; Blank, M.; Borchard, P. M.; Cauffman, S.
R.; Felch, K. L.; Weber, R. T.; Temkin, R. J.; Griffin, R. G.; Maas, W.
E. Phys. Chem. Chem. Phys. 2010, 12, 5850-5860.

Fig. 1. 1H SSNMR spectra of four cocrystals of the drug tenoxicam at obtained at a MAS spinning rate of 35 kHz and a static magnetic field strength of 16.4 T. A measurement temperature of 283 K was used. For details about the interpretation of these spectra, see {9}.
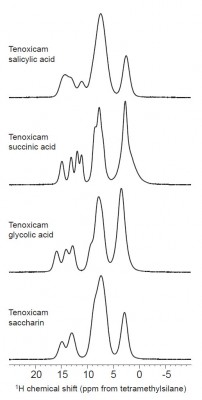
Fig. 2. 1H SSNMR spectra of amorphous quinapril hydrochloride (Sigma-Aldrich Co., St. Louis, MO, USA) obtained using conventional MAS and with DUMBO dipolar decoupling at a static field of 16.4 T. The MAS spectrum and DUMBO spectrum were obtained using MAS rates of 35 and 30 kHz, respectively. A measurement temperature of 283 K was used.
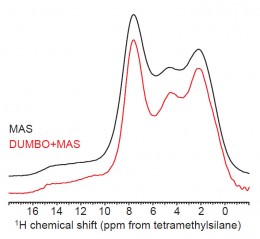
Fig. 3. 13C CP-MAS spectra obtained with DNP (microwaves on) and without DNP (microwaves off) of an amorphous solid dispersion containing 30% diflunisal in PVP {7}. The dispersion was impregnated with 16 mM of bCTbK radical (bis-TEMPO-bis-ketal, where TEMPO is (2,2,6,6-tetramethylpiperidin-1-yl)oxidanyl)) using 1,1,2,2-tetrachloroethane solvent (leading to signals in the 70-80 ppm region). Centerbands appear in the 180 to 10 ppm region. Each spectrum is the result of 16 scans. A relaxation delay of 7 s was used, during which DNP buildup occurs when microwave irradiation is applied. Spectra were obtained using a MAS rate of 8 kHz with a static field of 9.4 T. A measurement temperature of 100 K was used.