DNP for The Structural Study of Small Organic Molecules: Pharmaceutical Applications
About 80% of all active pharmaceutical ingredients (APIs) are administered as solids, the majority of which display polymorphism. During formulation, APIs are regularly exposed to invasive procedures (e.g. granulation, extrusion, milling, compaction, etc.) that can possibly induce polymorph transitions.
Because each polymorph is characterized by a unique set of physical and chemical properties, polymorph transitions can affect drug activity with implications in patenting rights. Accessing the structure of the API before, during and after formulation is hence of primary importance for pharmacy.
This article highlights recent progress of NMR in the atomic-level structural analysis of small powdered organic compounds used as APIs at natural isotopic abundance (NA) that were only made possible by the latest technological and theoretical advances in solid-state dynamic nuclear polarization (DNP) NMR.
DNP Sample Preparation Methods for Structural Investigations of APIs
The impact of DNP in the investigation of diluted – previously inaccessible – portions of solid materials (e.g. surfaces, chain ends, branching sites, defects, etc) became clear soon after the first commercial spectrometer for DNP was made available in 2010 by Bruker. However, the value of DNP for the structural investigation of pharmaceutical compounds only became apparent after appropriate sample preparation procedures allowing incorporation of the polarizing agent without altering the integrity of the solid sample were developed, such as the so-called incipient wetness impregnation [1] or methods based on spray-drying and hot-melt extrusion of amorphous solid dispersions [2]. The availability of reliable methods for incorporating the polarizing agent into the sample while avoiding sample disruption have since enabled the acquisition of NMR experiments that were impractical or even impossible on small organic molecules using standard NMR, opening new avenues for structural elucidation of APIs and solid formulations via DNP.
From Spatial Proximity to Internuclear Distances: “quantitative” DNP
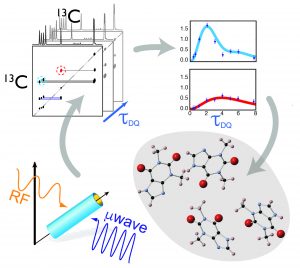
However, until a few years ago, applications of DNP to the study of pharmaceutical solids could mostly provide qualitative details about the structure of the API or the morphology of the solid formulation, typically expressed as spatial proximity between atoms. In a recent paper appeared in the Wiley journal Angewandte Chemie International Edition, a team of researchers at the University of Aix-Marseille showed for the first time that DNP can also enable the measurement of internuclear distances between carbon nuclei on NA APIs [3]. Specifically, the intensity of the correlation peaks appearing in a series of double quantum/single quantum (DQ/SQ) 13C-13C dipolar correlation spectra acquired via DNP using different dipolar evolution times provided quantitative structural constraints sensitive to carbon-carbon internuclear distances up to 6.5 Å. The authors proposed a straightforward method to relate these 13C NMR dipolar data to the crystal structure of the API, which does not rely on first principle calculations. The structural constraints obtained by DNP appear in the form of dipolar build-up curves (see Figure 1) determined by the set of internuclear distances in the solid structure. The build-up curves expected for a trial structure can be easily calculated from the knowledge of the atom positions in the structure. The structure providing the best matching with the experimental curves for all the analyzed correlation peaks is identified as the correct polymorph. The method was demonstrated on a sample of NA powdered theophylline, and proved extremely sensitive to both the molecular conformation of the API in the solid and to the presence of distinctive supramolecular motifs ubiquitous in solid APIs, like π-π stacking. Importantly, the acquisition of these spectra on NA theophylline took less than a weekend (instead of almost 20 years without DNP!), which was only made possible by the large absolute sensitivity enhancement of DNP.
Owing to the unique structural constraints obtained using DNP, trial crystal structures of the API theophylline could be rapidly and effectively discriminated. We anticipate that combining this method with powder X-ray diffraction, crystal-structure prediction, and first principles computation of NMR chemical shifts will pave the way to de novo 3D structure elucidation of pharmaceutical powders.
References:
- A.J. Rossini, A. Zagdoun, F. Hegner, M. Schwartzwälder, D. Gajan, C. Copéret, A. Lesage, L. Emsley, J. Am. Chem. Soc. 134, 16899 (2012)
- Q.Z. Ni, F. Yang, T.V. Can, I.V. Sergeyev, S.M. D’Addio, S.K. Jawla, Y. Li, M.P. Lipert, W. Xu, R.T. Williamson, A. Leone, R.G. Griffin, Y. Su, J. Phys. Chem B 121, 8132 (2017)
- G. Mollica, M. Dekhil, F. Ziarelli, P. Thureau, S. Viel, Angew. Chem. Int. Ed. 54, 6028 (2015)