timsTOF Pro 2

A quantum leap in 4D-Omics
The new standard for high speed, high sensitivity 4D-Multiomics
Liquid chromatography (LC) coupled with mass spectrometry (MS) has become the gold standard in various omics fields. However, the coverage of proteomes or metabolomes in complex biological samples remains challenging due to limited speed, sensitivity and resolution of current mass spectrometers.
Adding trapped ion mobility spectrometry (TIMS) to the equation unlocks the parallel accumulation serial fragmentation (PASEF®) acquisition method to provide extremely high MS/MS speed and sensitivity, requiring minimal sample amounts.
Dual-TIMS and CCS-enabled analysis
TIMS is first and foremost a separation technique in the gas phase. This resolves sample complexity through an added dimension of separation in addition to high performance liquid chromatography (HPLC) and mass spectrometry, increasing peak capacity and confidence in compound characterization.
Equally important, the TIMS device also accumulates and concentrates ions of a given m/z and mobility, enabling a unique increase in sensitivity and speed.
A near 100% duty cycle can be achieved with the dual-TIMS technology facilitating accumulation in the front section, while ions in the rear section are sequentially released depending on their mobility. This process of parallel accumulation serial fragmentation (PASEF®) enables collisional cross section (CCS) analysis.

CCS-enabled analysis opens up many further analytical possibilities, from greater certainty of compound identification to confident library matching and lower false discovery rates (FDRs) in large datasets.
PASEF®

The timsTOF Pro 2 comes with our innovative dual-TIMS analyzer that offers three times higher ion capacity. Simplified ion optics maximize ion transfer and sensitivity to set a new standard in 4D-Multiomics. PASEF® is a redesigned MS/MS technology to meet speed requirements of omics applications. Ions are separated using trapped ion mobility spectrometry, eluted (~ 100 ms) and detected in the quadrupole time of flight (QTOF), generating the TIMS MS heat map. PASEF® then uses the same TIMS separation to serially fragment ions. The quadrupole isolates a certain ion species during its elution for MS/MS and immediately shifts to the next precursor. Parent and fragment spectra are aligned by mobility values. The timsTOF Pro 2 can achieve a sequencing speed of > 120 Hz in PASEF® mode. Furthermore, the MS/MS spectra quality of low abundant analytes can be enriched by selecting them several times.
dia-PASEF® adding confidence to your identifications
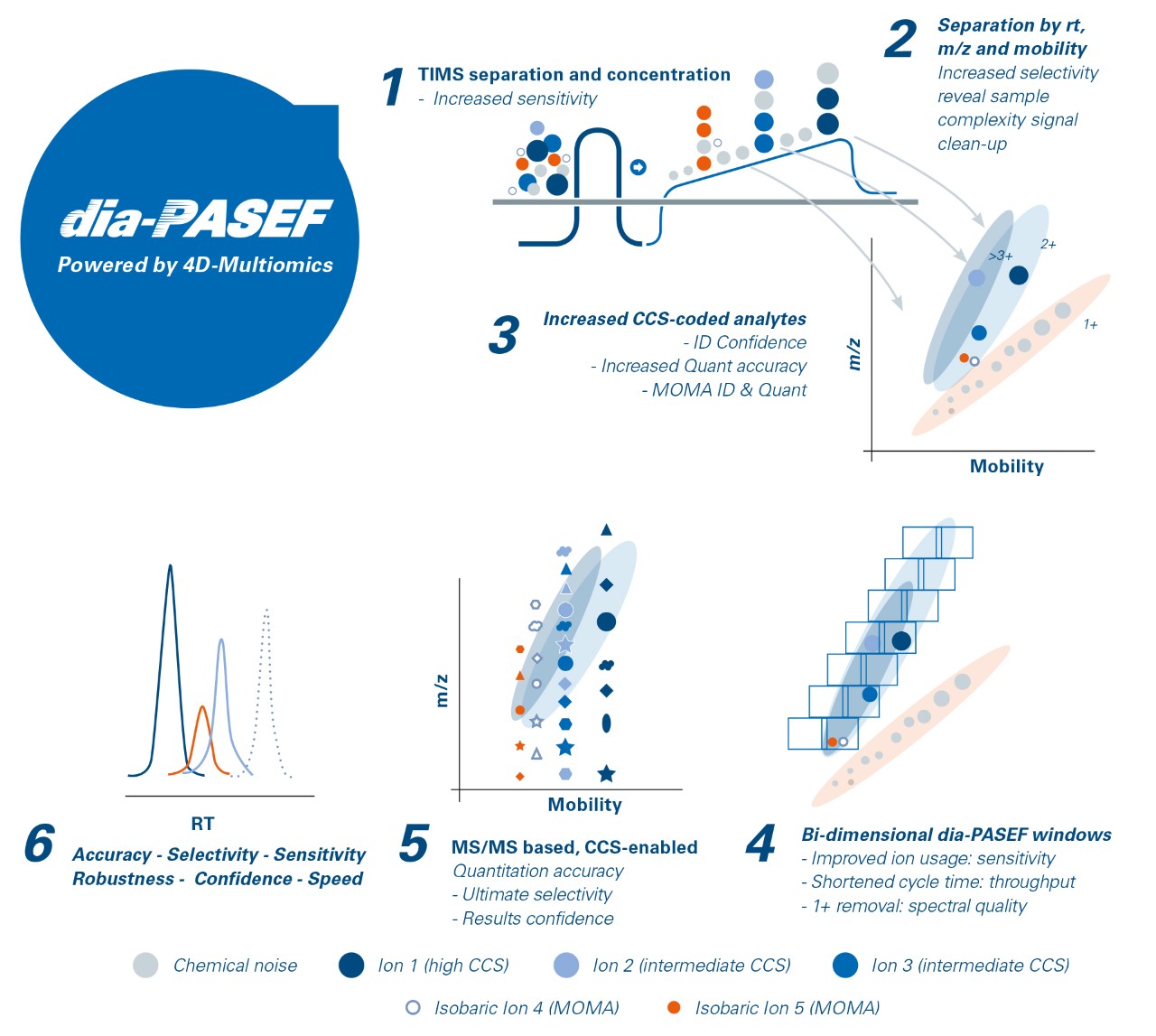
Data-independent acquisition (dia)-PASEF® is both more sensitive and selective than traditional DIA approaches as it applies the PASEF® principle to combine the advantages of DIA with the inherent ion efficiency of PASEF®. Over the entire liquid chromatography-mass spectrometry (LC-MS)/MS dia-PASEF® run, a perfect data cuboid is created containing m/z, ion mobility (CCS), retention time and intensity. TIMS separation increases selectivity, excludes singly charged precursors from fragmentation and cleans up the sample by concentrating signals from noise. Making use of the correlation of molecular weight and CCS coded information from the dual-TIMS funnel, dia-PASEF® enables highly confident identification.
Maximize peptide and protein sequence coverage
The robust stainless steel stacked ring ion guide (SRIG) configuration and optimized standard dia-PASEF® methods in timsTOF Pro 2 provides an unprecedented depth of proteome coverage in single shot proteomics. Nearly 9000 protein groups were identified from 200 ng of K562 lysate in 35 minute gradients (~30 sample per day) using a PepSep™ 25 cm column with nanoElute® on a timsTOF Pro 2 and a TIMScore™ powered spectral library.
The timsTOF Pro 2 provides in-depth proteome coverage for everyday cell line proteome quantification experiments directly by database searching and matching between runs without the need for a spectral library. Different database search strategies resulted in very comparable results. Bruker ProteoScape™, enabling real time protein identifications, and MaxQuant resulted in similar ID numbers at both protein and peptide level.

High-throughput targeted proteomics at unprecedented sensitivity
prm-PASEF® benefits from TIMS as a 4th dimension of separation for unmatched selectivity, the time-focusing of ions in the TIMS cartridge to increase sensitivity, and the high speed facilitated by PASEF® to increase the number of precursor targets. In a single acquisition several hundred precursors can be targeted without compromising the unique selectivity and high sensitivity of the timsTOF Pro 2 instrument.
Combining sensitivity, speed, and selectivity, the prm-PASEF® acquisition method delivered high reproducibility and accurate quantitation for either a high number of targets or for application with short chromatography gradients (i.e. <5 minutes).


"We’ve been working closely with Bruker to build prm-PASEF® from the ground-up. Throughout our collaboration, we’ve been impressed by the world-class combination of speed, sensitivity, and robustness provided by prm-PASEF®. We are excited to take this performance to the next level on the timsTOF Pro 2."
Jarrod A. Marto, Ph.D., Associate Professor, Department of Pathology, Brigham and Women’s Hospital and Harvard Medical School, Dana-Farber Cancer Institute, Boston, USA
Expanding the Horizons with 4D-Metabolomics™

Non-targeted metabolomics benefits from high MS/MS coverage as a key component of compound annotation. Our optimized 4D-Metabolomics™ methods provide both quantitative MS profiling data and MS/MS fragmentation spectra in the same sample analysis, achieving majority feature coverage even at low analyte concentrations. PASEF® provides mobility selection prior to fragmentation resulting in clean and unambiguous fragment spectra.
PASEF® is able to mobility select and fragment each compound separately, resulting in clean fragment spectra. Removal of matrix derived MS/MS fragments provides better library matching for Proadifen in MetaboScape® and hence higher confidence in results.
Higher confidence with 4D-Lipidomics™
Analogous to proteomics samples, lipid extracts have a high sample complexity caused by the structural diversity of lipids. High quality MS/MS spectra are integral to obtaining confident lipid annotations. PASEF® unlocks CCS-enabled workflows which can be used to additionally boost lipid annotation confidence.
Lipidomics data acquired on a timsTOF Pro 2 can be easily analyzed with MetaboScape®, which features a library-free annotation tool that utilizes published fragmentation rules and novel CCS prediction tools. Make full advantage of all 4 acquired dimensions.
PASEF® can fragment >10x more precursors by using mobility separation. This removes overlapping contaminants and resolves isobaric as well as isomeric lipids. The resulting MS/MS spectra show unique fragments for each lipid class resulting in annotations with increased confidence.



"PASEF® already out of the box increases the precursor coverage of lipids with an associated MS2 spectra to 70%. These MS2 spectra are essential for the correct identification of lipids. On top, the use of CCS adds additional confidence in annotation by adding an additional layer of information to RT, MS and MS/MS."
Dr. Michael Witting, Co-Head Metabolomics and Proteomics Core, Helmholtz Zentrum Munich, Neuherberg, Germany
Ultrahigh-throughput confident screening and quantification in complex matrices
The exceptional speed of the timsTOF Pro 2 system allows full unknown screening with either GC-APCI or UHPLC VIP-HESI data acquisition. In combination with TASQ® software, Bruker offers a complete solution from data acquisition to automated data analysis.

Applying mobility-filtering to extracted ion chromatograms increases sensitivity in complex matrices by removing interferences. This can clearly be seen in the trace for Thiacloprid in onion at a concentration of 1 ng/mL. The lower traces have been filtered at progressively narrower mobility ranges, allowing a perfect match to the TargetScreener database, as indicated by the green color of the MRSQC score.

Extremely high robustness
Minimal Cleaning Required
Many MS instruments used for omics applications require monthly cleaning when run 24 hours a day on large sample cohorts. Performance degradation is
noticeable even over shorter time periods. The superior robustness of the timsTOF Pro 2 means that the instrument can be run 24/7 over many weeks without noticeable loss of sensitivity or other performance metrics.

"Since we started to work with the timsTOF Pro 26 months ago in February 2019, we have run more than 25000 LCMS samples, of which about 5000 have been non-depleted plasma digests. We had virtually zero downtime so far."
Roman Fischer, Ph.D., Associate Professor in Clinical Proteomcs, Target Discovery Institute, University of Oxford, United Kingdom
timsTOF Pro 2 Related Information














For Research Use Only. Not for use in clinical diagnostic procedures.