Magnetic Resonance
What is Magnetic Resonance (MR)?
Magnetic resonance is a physical effect that is applied in electron paramagnetic resonance (EPR) spectrometers and nuclear magnetic resonance (NMR) to study matter at atomic level to return detailed information about molecular structure.
Magnetic resonance spectroscopy has found application in chemistry, biology, and materials science, serving both research and industrial purposes. For instance, it plays a crucial role in structural chemistry by helping to elucidate molecular configurations and dynamics, which are paramount in drug development and materials science.
Additionally, magnetic resonance imaging (MRI) and Time-Domain Nuclear Magnetic Resonance (TD NMR) are applications of magnetic resonance. MRI provides non-invasive, detailed images of internal organs and tissues, aiding in early disease detection and management. TD NMR offers insights into molecular structure, dynamics, and interactions, and is used in fields like pharmaceuticals, polymers, food industry, and environmental science for quality control and structural analysis.
Bruker, a leading supplier of EPR spectrometers, NMR spectrometers and preclinical MRI instruments, has continually advanced these technologies.
In the world of magnetic resonance, the constituents of atoms, when exposed to a magnetic field, become perturbed by an oscillating field and react by generating an electromagnetic signal. This process, when targeting atomic nuclei, is termed ‘nuclear magnetic resonance’ (NMR), while focusing on electrons leads to ‘electron spin resonance’ (ESR) or ‘electron paramagnetic resonance’ (EPR). ESR was first observed by Russian physicist Yevgeny Konstantinovich Zavoisky in 1944, marking the beginning of many important discoveries in this field. Two years later, NMR was independently observed in the United States by teams led by Felix Bloch and Edward Mills Purcell, a milestone that earned them the 1952 Nobel Prize in Physics, underscoring the profound impact of magnetic resonance on science and technology.
The Principle of Nuclear Magnetic Resonance (NMR) Spectroscopy
In NMR spectroscopy, samples for analysis are brought into a strong magnetic field. The atomic nuclei in the sample must have a particular quantum mechanical property, called ‘non-zero nuclear spin,’ to interact with the magnetic field in the spectrometer. Many important nuclei, most notably hydrogen, have this property and can be analyzed by NMR. When a sample substance is brought into the magnetic field of the NMR spectrometer, the nuclear spins in the sample align with the magnetic field. In an NMR spectrometer, the sample is surrounded by radiofrequency (RF) coils, which can then be used to apply one or more RF pulses to the sample. The RF pulses perturb the nuclear spins in the sample. The response to this perturbation is the production of an electromagnetic response in the RF coil, which contains information about the nuclei in the sample and their surroundings. This can be used to infer the structure of a molecule, for example. The resonance frequency of the nuclear spins depends on the magnitude of the magnetic field.
With the field strengths that are typically used in NMR, protons resonate at frequencies of several hundred Megahertz (MHz). Excitingly, this allows researchers to determine not just the types of atoms present but also their positions in a molecule, leading to insights into its three-dimensional structure.
Bruker has played a leading role in the development and commercialization of NMR spectrometry. Bruker was founded in 1960 in Karlsruhe, Germany, by a group including German physicist and entrepreneur Dr. Günther Laukien, who was one of the first to recognize the commercial potential of NMR technology. His vision propelled Bruker to the forefront of NMR innovation, constantly evolving with technological advances. NMR spectroscopy experienced ongoing breakthroughs.
Among the most notable technological achievements are the use of Fourier-Transform NMR (FT-NMR), the use of superconducting magnets with ever-increasing field strength, and multidimensional NMR. Today, NMR continues to be an invaluable tool in various scientific disciplines, providing unmatched precision in molecular analysis.

Fourier-Transform Nuclear Magnetic Resonance
In FT-NMR, the sample substance is exposed to a very short, relatively strong burst of RF energy, exciting all the spins in the sample simultaneously. This rapid excitation generates overlapping resonance signals which are recorded by the spectrometer. These signals are then subjected to a mathematical analysis called Fourier transformation (FT), which converts time domain data into frequency domain data which can be displayed in an easily interpretable spectrum. The method is distinguished by its ability to resolve complex molecular compositions, significantly improving workflow efficiency in analytical laboratories. An FT-NMR spectrometer typically generates a spectrum as shown in Figure 1, which can be used by scientists to infer the structure of molecules or detect impurities in mixtures. This capability is vital across numerous fields, including quality control, where it ensures product consistency, or drug development, where precise molecular analysis can lead to the creation of more effective pharmaceutical compounds. Additionally, FT-NMR is extensively used in chemical research, materials research, particularly in energy storage, and biology, where understanding the three-dimensional structure of molecules is crucial. Its versatility allows researchers to explore both static and dynamic chemical properties, immensely contributing to the advancement of fundamental sciences.
FT-NMR was pioneered in the 1960s by Swiss physicist Richard Ernst, who was awarded the 1991 Nobel Prize in Chemistry for his groundbreaking contributions to the development of high-resolution NMR spectroscopy methodology. To this day, FT-NMR spectroscopy remains the most widely used method, owed largely to its ability to provide high-resolution data swiftly and reliably.
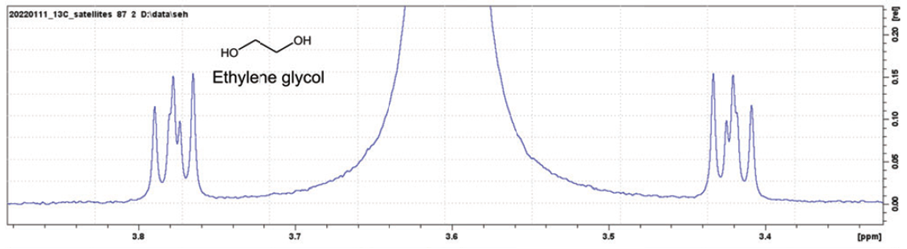
Stronger Magnetic Fields for Better Resolution

Using stronger magnetic fields improves NMR sensitivity and results in better spectral resolution. Today, in most NMR spectrometers, helium-cooled superconducting magnets generate the required strong static magnetic fields. Some of the strongest magnets in the world – with field strengths in the range of 30 Tesla – can be found in NMR spectrometers. In small and compact benchtop NMR spectrometers, permanent magnets are often used.
Multidimensional Nuclear Magnetic Resonance
Multidimensional NMR is a set of NMR methods, which uses special pulse sequences to yield data defined by more than one frequency axis, which enhances the resolution and the information content of an NMR spectrum. Multidimensional NMR spectroscopy forms the basis of determining the structure of biomolecules such as proteins, DNA or RNA, but is also routinely used in material science. Swiss physicist Kurt Wüthrich was awarded the 2002 Nobel Prize in Chemistry for his development of NMR spectroscopy to determine the three-dimensional structure of biological macromolecules in solution.
Learn More about NMR Spectroscopy:


Electron Paramagnetic Resonance (EPR) Spectroscopy
Electron Paramagnetic Resonance (EPR), also called Electron Spin Resonance (ESR), is a form of spectroscopy used to study molecules or atoms with unpaired electrons, including free radicals, transition metal ions, rare-earth ions, and defects in materials. Versatile and non-destructive, it can be used on solids, liquids, and gases. EPR is also the only technique that allows for unambiguous detection and quantification of unpaired electrons in samples, making an invaluable resource for both research and practical applications across a spectrum of scientific disciplines.
EPR Spectrometer Types
There are two primary types of Electron Paramagnetic Resonance (EPR) spectrometers: continuous wave (CW) and pulsed EPR, each offering unique approaches to analyzing samples with unpaired electrons.
In CW EPR, a sample is exposed to a constant frequency of microwaves while the magnetic field is varied. This allows for steady-state measurements of the sample's response, capturing the spectrum by observing changes in microwave reflection as the magnetic field fluctuates. This technique is valuable for identification of the source of unpaired electron spins in a sample and provides clues into the interaction of the electron spin with its environment, such as hyperfine couplings.
By contrast, pulsed EPR operates similarly to Nuclear Magnetic Resonance (NMR) techniques, where the sample is subjected to a series of microwave pulses that stimulate spin transitions. The electromagnetic responses to these pulses are then captured and analyzed either in the time domain or in the frequency domain using the Fourier transform. The main advantages in pulsed EPR lie in the access to information not easily attainable by conventional EPR, such as relaxation times, detailed hyperfine structure, and direct measurement of dipolar couplings. One can measure transient states in biomolecules that have spin labels to gain dynamical information or probe small molecule binding effects in protein sites.
The choice between using CW and pulsed EPR largely depends on specific research requirements, such as measurement precision and the type of information needed. The continuous advancement and innovation in EPR spectrometer technology have enhanced the flexibility and precision of EPR instruments, solidifying their role as invaluable tools in diverse fields like chemistry, materials science, and medicine, paving the way for further explorations and discoveries.
The Emergence of EPR Spectroscopy
Discovered by Yevgeny Zavoisky in 1944, EPR is similar to NMR but is based on exciting the spin of the electrons rather than the atomic nuclei. The frequencies of the electromagnetic signals in EPR are considerably higher than in NMR, typically in the range of tens to hundreds of gigahertz (GHz) or in the microwave band, therefore requiring microwave sources to excite the spins in the sample.
Commercial EPR instrumentation has evolved to keep pace with changing demands of new applications. Bruker entered the high field EPR market in the 1960s with the launch of the ER420 EPR spectrometer in collaboration with Prof. Weil, and quickly saw the need for a more intuitive, cost-effective and compact instrument – the first steps toward the significant development of an auto-tuning spectrometer for non-experts. In 1989 Bruker introduced the world’s first fully computer-controlled EPR spectrometer and, in 1992, the benchtop EMS104 was launched to support industrial applications.
Interest in developing a pulsed Fourier Transform (FT)-EPR spectrometer was led by Peter Hoefer, developer of the HYSCORE experiment, who later went on to win the Bruker Prize at the Royal Society of Chemistry in 2017.
Latest Developments in EPR Spectroscopy
The introduction of compact benchtop EPR spectrometer, Magnettech ESR5000 in 2019 has led to its application in a wide range of applications in food, medical devices and pharma. Bruker has also continued to innovate in high-end applications with the introduction of the ELEXSYS E780, the world's highest frequency commercial EPR spectrometer at 263 GHz. Bruker's technological advances also saw introduction of the Rapid Scan EPR accessory, an innovation that provides excellent time resolution for EPR (into the microsecond regime), improves signal-to noise, and decreases acquisition time.
A growing interest in materials and environmental science let to Bruker's development of high-end EPR instruments. The increased frequency offered byt the E780 provides the necessary g-factor resolution to differentiate between multiple paramagnetic species in a single sample. Information not previously available at lower frequencies becomes easily accessible providing crucial information on various chemical processes.
Applications of EPR
EPR is utilized by researchers worldwide across an extensive array of applications in biochemistry, biophysics, the development of new materials for electronics and energy storage, quantum information science, and environmental science.
Free radicals are often short-lived, yet they play crucial roles in many essential biological and chemical processes. Despite their transience, these highly reactive molecules can have a damaging impact on their surroundings, contributing to a range of diseases including cancer, Alzheimer’s, and cardiovascular disease, and are closely associated with aging processes. EPR spectroscopy's ability to accurately detect and quantify free radicals provides insights into the mechanisms of these processes, offering researchers valuable information for developing new approaches to combat related health disorders.
In biochemistry, EPR plays a pivotal role in elucidating the structure and quantification of small molecules by providing insights into free radical reactions and kinetics. On a broader molecular level, it assists in identifying metal centers, ligands, and substrate binding in macromolecules such as proteins, DNA, and RNA, while observing critical electron transfer pathways and amino acid-based radicals within proteins. In addition, through use of spin-labeling, Pulse EPR allows for probing inter-spin distances at separate sites in a biomolecule, providing insight into conformational changes and transient states in response to mechanistic events.
Quantum information science (QIS) has witnessed a resurgence in Pulse EPR technology due to the growing adoption of quantum mechanics by researchers in science and engineering. Numerous types of materials are being explored as quantum bits, or qubits, which are the basic unit of information in QIS. Pulse EPR has proven to be an essential technique in the study of molecular qubits based on electron spin due to direct access to spin properties, coherent control through microwave pulses, and even execution of quantum logic operations.
In the domain of electronics and energy storage, EPR reveals valuable insights into material behaviors under electrochemical conditions. It is particularly vital in battery development for detecting free radicals that induce electrode material degradation during charge and discharge cycles, thus enabling the creation of more efficient electrode materials and enhancing battery stability and efficiency. Defects in semiconductor materials have also gained traction in the materials research landscape due to their application in areas such as optoelectronics, photovoltaics, quantum computing and sensing. Identification of the defects, determination of the electronic
structure, monitoring catalytic processes, and more can be achieved using EPR spectroscopy.~In environmental science, EPR significantly contributes to sustainable energy development by characterizing systems with unpaired electrons, such as transition metal ions and radicals. This capability facilitates a deeper understanding of complex, multi-electron catalytic cycles, crucial for advancing future energy conversion technologies.

Learn More about EPR Spectroscopy:


Magnetic Resonance – A Versatile Analytical Technique
Building on years of expertise in magnetic resonance (MR) applications development and product innovation, Bruker provides cutting-edge instrumentation that caters to the world’s most advanced medical, life science, and materials science research. Beyond these high-end solutions, Bruker’s benchtop MR range is streamlining efficiency in quality control across applied and industrial markets.
This versatility makes MR an indispensable tool in key sectors such as environmental analysis, forensics, food quality assurance, and agriculture. The technology extends to a wide array of industrial applications, covering areas like chemicals, engineering, and manufacturing. In particular, industries dealing with plastics and polymers, pharmaceuticals and biopharmaceuticals, as well as energy, stand to benefit significantly from the precise and insightful data provided by MR technologies. This adaptability underscores the pivotal role of MR in promoting innovation and maintaining consistency across a diverse range of applications, showcasing Bruker’s commitment to advancing research and industrial efficiency through state-of-the-art magnetic resonance solutions.
Magnetic Resonance - Driving Progress in Diverse Applications
Life Science Research
In the rapidly changing fields of life science and pharmaceuticals, advanced research solutions are critical in accelerating discovery and expanding analytical capabilities to increase productivity. By leveraging tools like nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) spectroscopy, researchers can gain unprecedented insights into molecular structures and dynamics, ultimately enhancing drug development processes. These techniques offer precise data that is essential for understanding complex biological systems, facilitating the development of new therapies and personalized medicine approaches. The comprehensive data provided by MR technologies not only drives innovation but also optimizes laboratory workflows, allowing scientists to focus on developing novel treatments and interventions with increased efficacy and reduced timelines. Furthermore, with ongoing advancements in MR technology, researchers are equipped to tackle the most challenging questions in life science and pharmaceutical research, promising to revolutionize healthcare outcomes.
Materials Science Research
The materials science sector is evolving rapidly to drive the pace of industrial and technological change that significantly impacts the way we live. Innovations in high-tech materials are leading to groundbreaking advancements in various fields, such as the development of biomaterials that are crucial in medical science and healthcare. These materials are instrumental in creating state-of-the-art solutions for drug delivery systems that enhance the efficacy and safety of treatments. Nano-technology, another frontier in materials science, is playing a pivotal role in improving energy production and storage solutions, with a particular focus on battery development. This is crucial for supporting the growing demand for portable technology, ensuring that devices become more efficient, longer-lasting, and environmentally sustainable. Furthermore, the advancements in this sector are also bolstering sustainable practices across industries, paving the way for innovative applications that meet the pressing global challenges of today.

Industrial Applications
Bruker offers a wide range of testing and quality control solutions to help drive productivity across various industry sectors. MR spectroscopy is pivotal in mechanical and materials testing for industries such as aerospace and automotive, where precision and reliability are critical for safety and performance. In construction, MR applications ensure the durability and stability of materials, while in the chemical and coatings industries, they assist in analyzing compositions for optimal formulations and durability. The glass, ceramics, and metals sectors benefit from MR's insights into structural integrity and composition, aiding in advancements in manufacturing processes. For the cosmetics and electronics industries, MR spectroscopy offers a detailed analysis of product components, ensuring quality and compliance with industry standards. Furthermore, in the energy, gas, and power sectors, it plays a crucial role in monitoring material stability and performance under diverse conditions. The plastics and polymers industry leverages MR technology for developing materials with enhanced mechanical properties and longevity, which is equally applicable in the innovation of medical devices, where precise material characterization is essential for safety and efficacy.

Environmental
As environmental challenges mount, particularly with the rise of persistent organic pollutants (POPs) and microplastics, the need for sophisticated analytical solutions has become imperative. Magnetic Resonance (MR) technology stands out as a highly effective tool in addressing these concerns, providing precise methodologies for detecting and analyzing pollutants at trace levels. Bruker's MR solutions enable environmental scientists to conduct thorough assessments of pesticides, heavy metals, and organic contaminants across various media, including air, water, food, and soil. This level of detection is crucial for safeguarding ecosystems and public health, as it allows for the identification of pollutants before they can accumulate to hazardous levels. Furthermore, the versatility of MR technology supports its application across diverse environmental contexts, offering scalable solutions. By facilitating prompt and accurate contamination assessments, MR technology aids regulatory bodies and industries in implementing effective mitigation strategies, thus playing a vital role in environmental conservation efforts worldwide.

Forensics and Border Control
Magnetic Resonance (MR) technology offers a content-rich analytical method that provides invaluable insights by extracting extensive information from even the smallest samples in forensic science. This capability is particularly beneficial in forensic drug analysis, where MR techniques are adept at identifying new psychoactive substances (NPS), enhancing law enforcement's ability to tackle emerging drug threats. In the realm of criminal forensics, MR plays a crucial role by providing precise analyses of trace evidence, such as gunshot residues and glass fragments, which are instrumental in reconstructing criminal activities and identifying perpetrators. Furthermore, customs and border control authorities benefit from MR's efficiency in detecting illicit materials and contraband items, streamlining the monitoring processes at points of entry. The technique is also pivotal in art and document forgery investigations, as it helps authenticate materials by revealing minute discrepancies in composition and structure that might indicate falsification. By enabling such detailed forensic evaluations, MR technology supports the enforcement of legal frameworks, facilitating justice and ensuring public safety.

Food Quality Control
Bruker’s analytical solutions support quality control (QC) to uphold product quality and safety throughout the food chain, and ensure food authenticity to help prevent the threat of food fraud throughout the global food production industry, protecting consumers from adulterated and mislabeled products. In the ever-evolving marketplace, this level of scrutiny is essential as food safety concerns continually arise, demanding robust testing methods to identify contaminants and adulterants that could compromise consumer health. By implementing state-of-the-art magnetic resonance spectroscopy, manufacturers can detect even trace amounts of harmful substances with remarkable accuracy, ensuring that products meet rigorous safety standards. This technology also aids in verifying the origin and composition of food products, helping businesses maintain transparency and build trust with consumers who increasingly demand knowledge of the foods they consume. Consequently, such comprehensive validation processes not only protect public health but also uphold the integrity of global food supply chains, reinforcing consumer confidence in the brands they choose.

Agriculture and Animal Feed
Magnetic Resonance (MR) provides a sophisticated yet straightforward method for routine nutrient content testing that not only preserves the nutritional value of animal feed, pet food, and agricultural samples such as grains, seeds, and sugarcrops, but also plays a vital role in identifying potential toxins. This advanced analytical technique ensures that the nutritional requirements for animals are consistently met, thereby promoting optimal health and productivity. Additionally, by detecting contaminants and toxins early, MR helps in maintaining the safety and quality of agricultural products, making it an indispensable tool for compliance with stringent industry standards and regulations. The process involves precise MR spectroscopy, which delivers accurate and reliable data on nutrient composition and potential hazards, thereby assisting farmers and producers in making informed decisions regarding feed formulation and quality management. Implementing MR in nutritional testing not only aids in safeguarding animal health but also enhances the overall efficiency and sustainability of agricultural practices, ensuring that resources are used responsibly while maximizing output and maintaining ecological balance.

Bruker’s Commitment to High-Quality Research
Bruker has long been a pioneer in the field of magnetic resonance, with a steadfast commitment to pushing the boundaries of scientific knowledge. At the heart of Bruker's mission lies a dedication to advancing battery research and development, a crucial area that promises to enhance the performance and sustainability of energy storage solutions. Alongside this, Bruker excels in providing unparalleled sensitivity with Dynamic Nuclear Polarization-NMR (DNP-NMR) techniques, which offer insights into the molecular intricacies that drive advances in chemistry and materials science. Furthermore, Bruker places significant emphasis on functional and structural biology, utilizing their cutting-edge technology to unravel the complex mechanisms behind biological processes at a molecular level. In addition to these technological advancements, Bruker is devoted to global education, offering a range of expert training courses to empower scientists with the knowledge and skills necessary to harness the full potential of magnetic resonance technologies. This multifaceted approach not only supports innovation across numerous scientific disciplines but also ensures that researchers are well-equipped to make groundbreaking discoveries.
MR in Battery Research and Manufacturing
In the realm of battery research and manufacturing, magnetic resonance (MR) technologies are transforming the landscape with their ability to offer detailed molecular insights that drive innovation. MR plays a pivotal role by elucidating reaction mechanisms and degradation pathways that impact battery efficiency and longevity, offering a clearer understanding of how different materials interact at the molecular level. This powerful analytical capability is not only enhancing the design of safer and more reliable batteries but is also fostering the development of next-generation energy storage solutions. Furthermore, MR’s non-invasive nature allows for real-time monitoring of batteries under operational conditions, providing critical data that helps optimize performance and predict potential failures. As the demand for sustainable and high-performing energy storage solutions grows, MR's contributions to battery research are vital, supporting advancements that meet the mounting energy requirements of modern technology while promoting environmental sustainability.

Dynamic Nuclear Polarization (DNP) NMR
Dynamic Nuclear Polarization (DNP) NMR has emerged as a groundbreaking advancement in the realm of magnetic resonance, significantly enhancing signal sensitivity. This technique involves transferring polarization from electrons to nuclei, which results in a considerable increase in nuclear spin polarization, thereby magnifying the NMR signal by several orders of magnitude. DNP NMR is particularly beneficial in investigating complex molecular structures and dynamics at an unprecedented level of detail, opening new avenues for research in chemistry and materials science. Hyperpolarization through DNP not only speeds up data acquisition but also improves the quality of NMR spectra, allowing researchers to discern intricate molecular interactions that were previously challenging to detect. The integration of DNP with NMR technology marks a pivotal breakthrough, enabling a more comprehensive analysis of challenging samples and furthering the exploration of molecular mechanisms across various scientific disciplines.

NMR for Functional and Structural Biology
Functional and structural biology leverages advanced technologies like nuclear magnetic resonance (NMR) to delve deeply into the mechanisms of biological systems. Through NMR researchers can explore the dynamic behavior and three-dimensional structures of complex biomolecules, including proteins, nucleic acids, and membrane assemblies, crucial for understanding cellular processes. By revealing the interactions and conformational changes that underpin function, NMR provides a framework for analyzing the intricacies of molecular pathways involved in health and disease. This detailed molecular insight is not only vital for elucidating fundamental biological phenomena but also instrumental in the rational design of new therapeutic strategies. Moreover, the high resolution afforded by this spectroscopic method enables the accurate mapping of active sites and binding interactions, thereby optimizing drug targeting and efficacy. As structural biology continues to advance, the integration of this powerful technique with computational modeling and genomic data paves the way for personalized medicine approaches, enhancing the potential for tailored treatments and interventions.

MR in Education
Education is an integral component of Bruker's mission to advance scientific knowledge and innovation through magnetic resonance (MR) technology. By offering comprehensive training courses, Bruker ensures that researchers across various disciplines can harness the full potential of nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) systems. These educational programs are designed to accommodate both novices and experienced professionals, providing a robust foundation in fundamental theories while also exploring advanced analytical techniques. Participants gain hands-on experience with cutting-edge MR instruments, cultivating practical skills that propel their research capabilities. Furthermore, this initiative is not just about imparting technical knowledge; it fosters an environment of continuous learning and curiosity, encouraging researchers to apply MR technologies creatively in their work. Through education, Bruker supports the scientific community, equipping researchers to tackle modern challenges and make transformative discoveries across fields such as chemistry, biomedicine, and materials science.

World-Leading Nuclear Magnetic Resonance Spectroscopy
Bruker leads the world in the innovation, design, and manufacture of high-performance magnetic resonance instrumentation. Among their impressive portfolio is the world’s most powerful high-field 1.2 GHz NMR spectrometer, a cornerstone tool for advanced research applications that require precise and detailed molecular insights. Complementing this high-end machinery is their range of compact and cost-effective NMR benchtop instruments, specifically engineered for routine quality control and product testing applications. This versatility caters to both cutting-edge academic research and practical industrial use, demonstrating Bruker's commitment to delivering scalable solutions. Additionally, Bruker remains at the forefront of sustainability with their innovative, cryogen-free instruments, which utilize state-of-the-art magnet technology that minimizes environmental impact. Enhancing their versatility further, Bruker offers a wide variety of liquid and cryo probes, which are essential for adapting to numerous applications, spanning from intensive research endeavors to routine analytical tasks. Their comprehensive suite of tools not only advances scientific capabilities but also paves the way for more sustainable practices in scientific inquiry and industrial production.
Advanced Electron Paramagnetic Resonance Solutions
With a commitment to pushing the boundaries of scientific innovation, Bruker’s advanced pulse EPR spectroscopy technology supports cutting-edge research across the globe. These sophisticated tools empower researchers to delve into complex molecular phenomena with greater precision, significantly enhancing the potential for scientific breakthroughs. Bruker‘s advanced Fourier transform spectrometers are a crucial asset, offering high-frequency EPR capabilities at 94 GHz and 263 GHz. These frequencies open new avenues for exploring material properties and electron dynamics that were previously inaccessible, facilitating strides in fields such as chemistry, physics, and materials science. Furthermore, the introduction of new benchtop EPR instruments marks a notable advancement by increasing accessibility to this leading technique, making it feasible for laboratories of all sizes to harness the power of EPR spectroscopy. By democratizing access to advanced spectroscopic capabilities, Bruker not only supports global research initiatives but also fosters innovation across a diverse array of scientific disciplines, from pharmaceutical development to materials engineering, ultimately driving new discoveries and applications.
Best-in-Class Nuclear Magnetic Resonance and
Electron Paramagnetic Resonance Software
Bruker’s comprehensive software portfolio is designed to ensure rapid and reproducible NMR data acquisition alongside accurate results interpretation, enhancing precision and efficiency in research workflows. Central to this portfolio is the industry-leading TopSpin software, a tool that facilitates the transition to automated data acquisition and analysis, delivering results in an intuitive and visual format, thus simplifying the data handling process for researchers. Additionally, the AutoCalibrate software tool is an invaluable asset, continuously monitoring the system's health to guarantee long-term reliable performance of the NMR equipment, while SmartDriveNMR brings advanced data acquisition capabilities to open access NMR environments, broadening accessibility and operational ease. In collaboration with Mestrelab Research, a recognized leader in NMR software, Bruker offers a fully integrated digital approach, streamlining operations and enhancing connectivity across research platforms. Moreover, effective electron paramagnetic resonance (EPR) spectroscopy hinges on efficient data collection and precise processing, which Bruker’s dedicated software suites adeptly handle, ensuring reliable EPR data acquisition and analysis for continuous wave EPR, pulsed EPR, and EPR imaging. These robust software solutions optimize the research experience, enabling scientists to harness the full potential of EPR technologies across diverse applications.
How Magnetic Resonance is Aiding Customer Research
Bruker partners and collaborates with customers across the globe who are at the forefront of groundbreaking research. By leveraging the advanced capabilities of Bruker NMR systems, these researchers are uncovering novel insights that push the boundaries of scientific understanding. In the realm of food authentication, Bruker NMR aids in precisely determining the authenticity and quality of food products, helping to combat the global issue of food fraud and ensuring consumer safety. Furthermore, the technology plays a pivotal role in the study of COVID-19, where researchers utilize it to analyze virus-host interactions and accelerate the development of therapeutic strategies. In the field of sustainable energy, NMR systems provide critical data on new materials, aiding in the development of next-generation energy storage solutions that promise enhanced efficiency and environmental sustainability. Additionally, in the treatment of diseases, NMR spectroscopy offers unparalleled insights into the molecular pathways of various conditions, facilitating the design of more effective and targeted therapies. This collaborative approach not only reinforces Bruker's impact on cutting-edge research but also fosters innovation across a diverse range of scientific endeavors.

Enhance your Knowledge of Magnetic Resonance Techniques
Bruker is committed to empowering researchers using nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) to advance science by offering a range of comprehensive training courses and education programs. The in-person and online courses are led by our team of magnetic resonance experts and provide detailed explanations helping scientists to master NMR and EPR techniques at all skill levels. Tailored to both beginners and seasoned professionals, these courses cover foundational theories, advanced analytical techniques, and the latest advancements in MR technologies. Participants gain hands-on experience with state-of-the-art MR instruments, thereby enhancing their practical skills and confidence in conducting independent research. By integrating theoretical instruction with practical application, Bruker ensures that attendees are equipped to leverage MR technologies in their respective fields, fostering innovation and accelerating progress in science and industry. Through these educational efforts, Bruker not only supports knowledge enhancement but also prepares researchers to tackle the complex challenges of modern scientific inquiries effectively.
NMR training courses and education programs
Bruker’s NMR training courses offer researchers a comprehensive suite of educational opportunities that span from the fundamentals of nuclear magnetic resonance to the mastery of advanced analytical techniques. These meticulously crafted courses are designed to impart a deep understanding of NMR theory, equipping participants with the knowledge needed to utilize their systems effectively and innovatively. With a rich curriculum that delves into various application areas, attendees are guided by Bruker’s experienced trainers who bring extensive expertise and insights into the classroom. This dynamic learning environment ensures that researchers receive personalized guidance tailored to their specific research needs, enabling them to maximize the potential of their NMR equipment and software. Whether targeting applications in chemistry, materials science, or other scientific disciplines, Bruker’s educational programs help scientists leverage NMR technology to push the boundaries of discovery and innovation.
EPR training courses and education programs
With Bruker’s online and in-person EPR training programs, both beginners and advanced users can learn the skills and information needed to use EPR systems to their full potential. These educational courses cover a wide range of EPR applications from materials science to biological systems.








